

Technique et protocole d'étude
A1. Techniques et protocoles d’étude
A1.1. Séparation et tri des cristaux d’apatite
La technique d’extraction des apatites passe par une série d’étapes de préparation de la roche brute qui est indispensable afin de récupérer et concentrer la fraction minérale lourde et non magnétique contenant les cristaux d’apatite. Les étapes de préparation des échantillons pour l’extraction des apatites sont synthétisées sur la Figure A1-1.

A1.1.1. Broyage et tamisage
Deux à cinq kilos de roches sont prélevés par échantillon. L’apatite est un minéral particulièrement sensible à l’altération (Braun et al., 1994) ; la collecte a préférentiellement été réalisée pour des roches récemment mises à l’affleurement ou sur des surfaces saines. Le volume d’échantillon à broyer est fonction du type de lithologie collectée car la probabilité de récolter des cristaux d’apatite est variable.
Les échantillons sont débités par un broyeur à mâchoires en petits fragments de la taille d’un fin gravier. La roche est broyée à plusieurs reprises avec un écartement des mâchoires de plus en plus réduit afin d’obtenir une granulométrie plus fine à chaque étape.
Les éléments broyés sont tamisés dans trois séries de tamis en métal avec une trame de 400, 200 et 63 μm. La fraction inférieure à 400 μm est retirée entre chaque passage au broyeur afin de ne pas réduire en poudre les grains déjà séparés. Le tamisage est ensuite effectué avec un tamis à 200 μm. La fraction inférieure à 200 μm est tamisée à 63 μm sous l’eau courante, permettant d’extraire les fractions fines. La fraction granulométrique retenue est comprise entre 200 et 63 µm. Les minéraux de trop petite taille ne peuvent pas être utilisés de manière efficace pour le comptage des traces de fission et pour l’analyse (U-Th)/He. Pour des cristaux dépassant une granulométrie de 400 µm, de nombreuses inclusions et fractures vont perturber les analyses et ces cristaux ne seront pas exploitables.
A1.1.2. Séparation densimétrique et magnétique
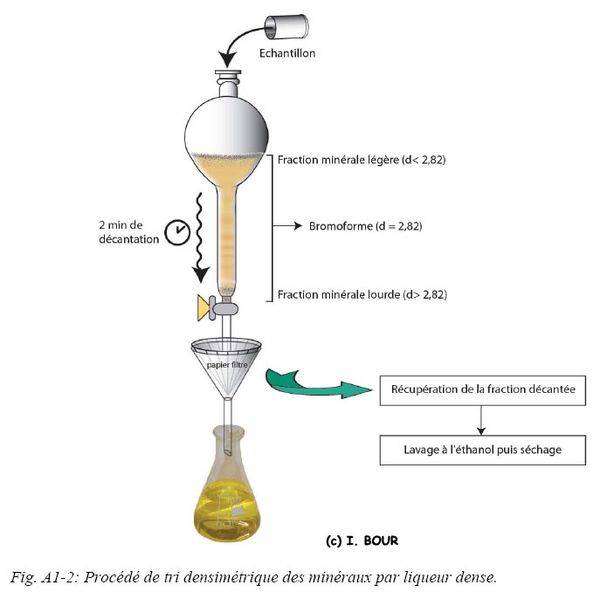
L’apatite est un minéral de forte densité comprise entre 3,1 et 3,3. Afin de séparer les différents minéraux présents dans la fraction minérale fine [200-63] µm, le procédé de séparation par liqueur dense (ou séparation par flottaison) est nécessaire. La méthode densimétrique au tétrabromoéthane ou plus communément appelé bromoforme a été utilisée. Cette liqueur dense possédant une densité de 2,82, permet de décanter la fraction minérale lourde supérieure à une densité de 2,82. La séparation s’effectue sous hotte aspirante dans une ampoule à décanter d’un litre où le sable est introduit jusqu’à former un dépôt d’environ un à deux centimètres sur le bromoforme. Une agitation pendant une minute suivie d’une phase de décantation d’une à deux minutes permet d’obtenir une bonne séparation. La fraction de densité supérieure à 2,82 est récupérée à la base de l’ampoule (Fig. A1-2), tandis que la fraction plus légère (<2,82) flotte sur le bromoforme.
Les fractions sont ensuite lavées à l’éthanol afin de les nettoyer (solubilisation et récupération du bromoforme).
Selon les échantillons, les cristaux de zircon peuvent être plus abondants par rapport aux cristaux apatite. Pour limiter la confusion entre les deux minéraux et afin de gagner du temps lors de l’étape de tri au microscope, une nouvelle séparation par liqueur dense peut être pratiquée. Elle peut se réaliser avec le diodométhane (d=3,38) selon le même principe décrit précédemment. Avec une densité plus importante que le bromoforme, le diodométhane permettra de séparer l’apatite (plus légère) du zircon (de densité plus lourde : 4,2 à 4,5). Dans ce dernier cas, seule la fraction légère sera récupérée pour l’observation.
Le rinçage de la fraction minérale est réalisé avec de l’acétone.
Une séparation magnétique sur la fraction minérale récupérée de l’étape précédente est réalisée afin de soustraire les minéraux magnétiques, notamment la magnétite et la biotite ainsi que les autres minéraux légèrement ferromagnétiques. Cette séparation est faite à partir d’un séparateur à barrière de champ de type Frantz® (Fig. A1-3). Cette séparation magnétique s’opère à l’aide d’un puissant électroaimant ventouse utilisant un courant avec une intensité de 1 A. Les grains circulent dans un conduit à débit régulier cerné par l’aimant. Les minéraux soumis au champ magnétique empreintent deux trajectoires différentes selon leur magnétisme, ce qui permet leur séparation en deux lots. Le séparateur Frantz® présente l’avantage de combiner la force de pesanteur et également d’attraction. La fraction non magnétique est essentiellement reconnaissable par sa couleur claire. C’est cette dernière qui sera récupérée pour la sélection optique.

A1.1.3. Critères d’identification des cristaux d’apatite
Les apatites sont des minéraux accessoires dont la reconnaissance est plus ou moins aisée selon leur état de préservation (forme, cassure, couleur). L’origine étymologique, venant du grec (apatan) signifierait tromper, d’où leur nom (Werner, 1786). Il est par ailleurs facile de les confondre avec d’autres minéraux. Néanmoins, les apatites présentent un certain nombre de caractéristiques visuelles qui permettent de les identifier.
Parmi la fraction minérale lourde issue de la séparation densimétrique et magnétique, il subsiste malgré tout des minéraux que nous n’utiliserons pas (pyrite, quartz, oxydes, zircon). Un tri manuel réalisé à la loupe binoculaire polarisante de la fraction minérale est nécessaire.
L’apatite cristallise dans le système hexagonal et apparaît sous forme d’un hexagone (parfois aplati) plus ou moins allongé (en prisme ou en tablette) dont trois de ses facettes sont visibles lorsqu’il est disposé parallèlement à l’axe cristallographique c (Fig. A1-4.B et Fig. A1-5.A). Le minéral est automorphe avec des bordures pouvant être plus ou moins émoussées selon le degré de préservation. Ses facettes sont lisses contrairement aux autres minéraux de la paragénèse qui présentent des faces issues de plan de cassure.
En lumière naturelle au microscope, les apatites couvrent une gamme de couleurs diverses allant le plus souvent de l’incolore au jaune ocre. Des tons rosâtres à violacés peuvent être observés. Ces nuances colorimétriques proviennent de la concentration plus ou moins importante en manganèse (coloration jaune) ou en cérium (coloration vers le rosé ou le violacé). L’apatite est semi-transparente à translucide et son éclat peut être vitreux ou gras. Dans le cas d’une lame mince, son relief est légèrement plus élevé que celui du quartz. En lumière analysée, le minéral polarise dans les biréfringences du deuxième ordre (teintes dans les jaunes-bleutés).

’apatite peut être confondue dans certains cas avec d’autres minéraux partageant la paragénèse dont le zircon (ZrSiO4) notamment (Fig. A1-4.A). En effet, ce dernier est un minéral automorphe de morphologie et de couleur relativement voisines au premier abord. Il cristallise dans le système quadratique et ne présente par conséquent qu’une seule face à l’observateur (Fig. A1-5.B1). Il existe des cas de troncatures des faces du zircon pouvant faire apparaître une ligne de pente entre deux faces, imitant ainsi le système hexagonal (Fig. A1-5.B2).

Le zircon est souvent dans les nuances jaunes à rosâtres voire rougeâtres liées à une forte concentration en uranium et en thorium qui dégrade la structure cristalline. La coloration du zircon n’est pas un critère fiable pour sa reconnaissance car il peut être incolore. Son éclat adamantin est accompagné d’une brillance ainsi qu’une opacité plus importantes que l’apatite. Le zircon est un minéral de forme souvent allongée mais pouvant être trapue. Sa principale caractéristique réside au niveau de ses deux terminaisons axiales en pointes au niveau de son cristal. En général, le zircon est de taille plus réduite que l’apatite (~60 µm). Certaines apatites peuvent contenir des inclusions de zircon. Ces apatites seront à exclure de l’analyse, notamment pour la datation (U-Th)/He.
A1.1.4. Sélection des cristaux d’apatite
La sélection des cristaux d’apatite ne demande pas d’exigence particulière pour le comptage et les mesures des traces de fission. Les cristaux doivent être de taille supérieure à 50 µm, notamment en terme d’épaisseur (Fig. A1-6), pour limiter la perte de cristaux au polissage et maximiser l’aire d’observation à la surface de ces mêmes cristaux.

Pour la méthode (U-Th)/He, les exigences de sélection des grains d’apatite suivent un protocole plus délicat et contraignant. Le tri des grains d’apatite est effectué selon deux étapes.
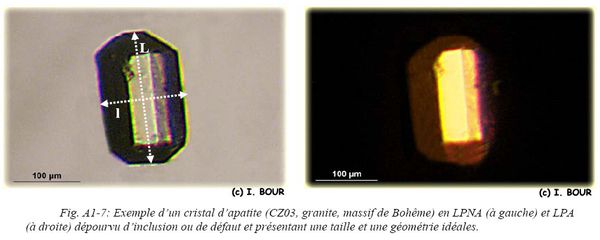
Une première étape de tri est basée sur le critère de taille et de forme cristalline (automorphe, allongé, trapu). Cette étape est effectuée à la loupe binoculaire en lumière naturelle transmise. Les grains d'apatite doivent posséder une géométrie particulière et des arrêtes nettes (Fig. A1-7) afin de déterminer leur taille ainsi que leur rapport surface/volume. Les cristaux d'apatite doivent être d’une longueur supérieure à 80 µm afin que la détermination du FT soit valable (acquisition d’une bonne précision).
La pureté des cristaux rentre en compte au cours d’une seconde phase de sélection. L’absence de fracture et l’absence totale d’inclusion sont également les critères de sélection indispensables pour les analyses par la méthode (U-Th)/He. L’apatite contient souvent des inclusions de zircon, monazite ou de verre pour les roches d’origine volcanique, de forme et de taille variables (de l’ordre de <1 à 10 µm) qui peuvent être riches en U et Th. La probabilité de présence des inclusions augmente avec la taille des apatites et dépend également de la chimie de la roche. En lumière polarisée, les inclusions apparaissent sous l’aspect de spots lumineux (Fig. A1-8) dont la biréfringence diffère par rapport au minéral hôte en raison d’une différence de système cristallin. Afin d’affiner le contrôle de l’absence d’inclusion, la présence des inclusions est également vérifiée en observant les grains dans une goutte d’alcool pour effectuer une observation sous un même plan focal.

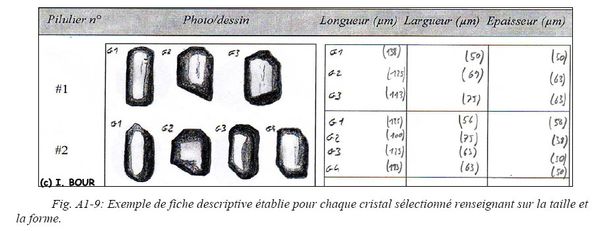
Une fiche descriptive de chaque grain sélectionné est ainsi établie pour l’analyse (U-Th)/He (Fig. A1-9) et sera nécessaire pour déterminer le facteur de correction FT (cf. II.2.3.4.).
A1.2. Protocole de préparation des échantillons
A1.2.1. Montage et polissage des cristaux
Chaque cristal d’apatite est prélevé à la loupe, grain par grain, à l’aide d’une aiguille puis fixé sur une coupelle par une colle d’araldite. Les cristaux d’apatite sont regroupés et sont rangés en lignes et en colonnes. Les cristaux sont disposés parallèlement à leur axe cristallographique c. La forme du cristal permet de repérer la direction de l’axe c qui est parallèle à la plus grande longueur du cristal. Organiser les cristaux de cette manière apporte un meilleur confort d’observation (localisation des cristaux plus aisée et plus rapide) mais cette organisation est également indispensable pour homogénéiser l’orientation des cristaux et donc les mesures ultérieures des densités et des longueurs de traces. L’ajout d’une pièce cylindrique sur la coupelle permet ainsi de constituer un moule récipient de 25 mm de diamètre. Après séchage de la colle d’araldite (durant une journée), le moule est rempli de résine synthétique d’époxy afin de constituer un plot pour chaque échantillon (Fig. A1-10). Les moules sont mis sous étuve à 40°C pendant une demi-journée afin que la résine se solidifie.

Après démoulage, la face sur laquelle se situent les cristaux (Fig. A1-11.A) est délicatement polie. Le polissage met en surface les cristaux d’apatite piégés dans la résine mais génère également une surface interne (Fig. A1-11.B) sur ces mêmes cristaux. Les irrégularités de surface sur les plots vont par la même occasion être supprimées. Le polissage des plots s’effectue selon différents paliers abrasifs (papiers de verre de 600 et 1200) de manière à éviter la perte des cristaux les plus petits. La finition du polissage (lustrage) est assurée avec des draps de polissage imbibés d’une pâte de diamant de 3 puis de 1µm. La qualité du polissage (absence de rayure d’abrasion, surface parfaitement plane,…) est contrôlée au microscope en lumière réfléchie.

La surface polie doit être traité par une solution acide afin que les traces de fission spontanées soient visibles au microscope optique. Cette étape est détaillée plus en amont (cf. II.1.3.1.).
A1.2.2. Irradiation neutronique
Les échantillons nécessitent une préparation préalable pour l’irradiation neutronique.
Les plots de résine contenant les échantillons sont préalablement recoupés sous forme de pastille rectangulaire de 2 mm d’épaisseur et sont ajustés à la dimension du conteneur d’irradiation. Chaque préparation a été surmontée d’une feuille de muscovite prédécoupée (détecteur externe) fixée à l’aide de ruban adhésif. Cette dernière est étroitement mise en contact avec la face polie de la pastille. Pendant la durée d’irradiation, le détecteur reçoit les fragments de fission des atomes d’235U provenant de la surface polie de la pastille qui lui fait face. Il en résulte, sur le détecteur, une image miroir des traces induites.
Afin de déterminer la fluence en neutrons (Φ) qui traversera l’épaisseur de l’ensemble des échantillons, trois verres dosimètre (Fig. A12) sont placés dans le conteneur selon trois positions (base, centre, sommet). Les dosimètres utilisés sont des pastilles de verres possédant une composition en uranium connue. Il en existe différentes catégories possédant des teneurs en uranium variables. Le verre Corning CN5 7146 à 12 ppm en uranium est le dosimètre employé pour cette étude. Le CN5 possède un rapport Th/U faible et permet d’obtenir dans le cas pratique une population de traces induites générées uniquement par la fission de l’uranium. L’emploi des trois dosimètres est indispensable pour calculer le flux de neutrons reçu par chaque échantillon. Les verres dosimètres constituent des indicateurs sur la variation du flux de neutrons à travers la succession des échantillons.
Les pastilles ainsi préparées avec leur détecteur sont empilées les unes sur les autres et sont introduites dans le conteneur (navette : Fig. A1-12). L’irradiation est effectuée sur une trentaine de pastilles par conteneur. Le matériel à irradier doit être nettoyé à l’alcool et assemblé avec des gants afin d’éliminer tout risque de dépôt de sels organiques (l’activité radiogénique en serait alors amplifiée rendant l’échantillon radioactif pendant une période de temps plus longue). Après irradiation, un mois est nécessaire afin de réduire l’activité des échantillons à un seuil équivalent au bruit de fond naturel.

Les traces de fission induites générées par l’irradiation neutronique sont enregistrés sont les feuilles de micas. La révélation des ces traces s’effectuent également par traitement acide dont les conditions sont décrit en II.1.3.2.
A1.2.3. Préparations de la lame mince
Chaque pastille échantillon est fixée sur une lame de verre côte à côte avec son détecteur externe respectif (Fig. A1-13). Le détecteur est au préalable collé sur une pastille de verre pour limiter les différences importantes d’épaisseur entre le détecteur et l’échantillon (mise sur un même plan focal). Un vernis à ongle est suffisant pour coller les échantillons et leurs détecteurs. En cas d’erreur de positionnement, un bain dans l’acétone permet de décoller les différents éléments fixés sur la lame. Les lames d’étude sont toutes organisées de la même manière car les observations en microscopie qui vont suivre seront effectuées sur une platine mobile assistée par un logiciel de positionnement.

A1.3 Protocoles d’analyses (U-Th)/He
A1.3.1. Montage des cristaux
Les grains sélectionnés doivent être placés dans une capsule de platine cylindrique d’1 mm de diamètre. Le conditionnement des grains d’apatite dans une capsule de platine permet leur transport et leur maniement pour l’analyse en spectrométrie. Compte tenu de la transparence des cristaux, le conditionnement des cristaux dans une capsule de platine permet d’homogénéiser la température de chauffe dans celle-ci.
Dans la probabilité où la teneur en 4He serait faible ou dans le cas de cristaux limités en taille, plusieurs grains peuvent être introduits dans une même capsule. Dans cette situation, des cristaux de même caractéristique géométrique et de taille voisine ou de même β doivent être mis ensemble dans la capsule. Celle-ci est fermée non hermétiquement à ses extrémités de manière à empêcher la perte des grains mais laissant suffisamment d’ouverture pour le dégazage de l’hélium lors de son extraction et pour la dissolution chimique des cristaux.
A1.3.2. Mesure de la concentration d’hélium
A1.3.2.1. Extraction-purification des gaz
Le système d’extraction-purification se présente sous la forme d’une ligne en acier inoxydable offrant un blanc d’hélium (teneur correspondant au bruit de fond) très faible. L’installation est équipée par de multiples éléments mécaniques, ainsi qu’un système de purification et d’analyse schématisés et détaillés ci-dessous (Fig. A1-14 et Fig. A1-15).

Les échantillons sont placés sur un support en cuivre (Fig. A1-15) au sein d’une enceinte mise sous un vide poussé atteignant les 10-9 mbar. Les capsules sont disposées de manière identique (à l’horizontale sans inclinaison) sur le portoir de l’enceinte d’extraction afin que le processus de chauffage au laser soit homogène et atteigne la température fixée.
L’enceinte est fermée par une fenêtre en saphir surmontée par un laser fibré dopé à l’Ytterbium possédant une puissance maximale de 10 W. L’extraction de l’hélium contenu dans les cristaux d’apatite est réalisée par chauffage via le rayon incident du laser. Celui-ci est placé à la distance focale de 4 cm au dessus de l’échantillon. La fenêtre en saphir de l’enceinte à l’avantage de posséder un blanc d’hélium très faible et présente l’intérêt d’être transparente pour l’intégralité du spectre lumineux qui compose le faisceau laser (1,01 µm de longueur d’onde dans le domaine de l’infrarouge).

La température de chauffage est contrôlée en temps réel à partir d’une caméra CCD (Fig. A1-16) qui photographie par intervalle de temps régulier la capsule chauffée. Un paramétrage de la température de chauffage est réalisé par comparaison avec la couleur d’un corps noir (éclat qui est fonction de la température). L’ampérage du laser est modulé afin d’être calé sur la température de chauffe de l’ordre de 1050°C pendant 5 minutes (Fig. A1-16). Le dégazage par chauffage au laser permet d’obtenir un blanc quasi identique par rapport à un blanc réalisé à froid.

La phase gazeuse présente dans le système est purifiée au moyen d’un piège à charbon actif refroidi à l’azote liquide (-180°C) permettant d’absorber principalement l’Ar et le CO2. Le SAES Getter constitue un autre type de piège gazeux capturant essentiellement l’hydrogène par adsorption.
A1.3.2.2. Analyse de l’hélium
Le gaz purifié est mesuré par un spectromètre quadrupôlaire (Prisma QMS 100). Le Quadrupôle est muni d’un dispositif qui ionise et accélère les gaz puis les focalise en direction des détecteurs selon leur rapport masse/charge (Fig. A1-17). Les détecteurs permettent de mesurer l’intensité du courant généré par chaque faisceau d’ions isolés issus du gaz (multicollection). Au niveau de l’analyseur, les atomes ionisés et accélérés, vont déposer leurs charges sur les détecteurs et induire un courant dont l’ampérage sera fonction de la concentration en gaz. Cette mesure de l’intensité traduit l’abondance relative de chaque isotope présent dans le système.

L’emploi d’un standard va permettre de calibrer le capteur. Celui-ci va fournir une valeur d’ampérage correspondant à la concentration connue du standard (conversion du signal électrique en valeur de concentration). Un volume défini (~1 cm3) d’un standard gazeux interne possédant un rapport 4He/3He connu est mesuré tous les quatre échantillons afin de contrôler la reproductibilité et stabilité des mesures. La source mesurée n’est pas toujours reproductible d’un jour à l’autre (dérive analytique).
Une quantité connue d’3He (spike) est mélangée avec l’4He extrait de l’échantillon afin de déterminer le rapport 4He/3He. L’ajout du spike 3He est nécessaire pour retranscrire un signal (en ampères) en nombre de mol.
La mesure d’un blanc est réalisée après chaque remplacement d’échantillons (ouverture de la ligne). La mesure du blanc d’He est nécessaire (analyse sans échantillon) afin de soustraire la valeur de ce dernier aux mesures des teneurs d’He de l’échantillon. Le blanc variera en fonction des techniques employées.
Pour chaque échantillon, un deuxième dégazage réalisé dans les mêmes conditions que le premier et une nouvelle analyse de la réextraction sont nécessaires afin de contrôler que l’4He a bien été entièrement extrait lors du premier palier de chauffe. Lors de la deuxième séquence de chauffage, la mesure d’une quantité d’4He supérieure à la valeur seuil du blanc est en général la conséquence de la présence d’inclusions dans les cristaux (zircon, monazite) riches en U et Th. Si la seconde réextraction présente une valeur en 4He similaire au blanc ou de l’erreur analytique associée, il n’y a alors pas de réextraction significative ce qui permet de s'assurer du dégazage complet de l'échantillon lors de la première séquence de chauffage. Les analyses du second dégazage présentant un écart significatif par rapport au blanc ne vont pas nous fournir un âge exploitable et devront être exclues.
A1.3.3. Mesure de la concentration d’uranium et de thorium
Un autre protocole de préparation des échantillons est réalisé pour la mesure de leur concentration en uranium et en thorium.
A1.3.3.1.Dilution chimique des cristaux d’apatite
L’analyse ICP-MS est réalisée sur des sources liquides, il est donc nécessaire d’effectuer une dissolution des échantillons. Les capsules contenant les cristaux d’apatite dégazés, sont récupérées et insérées dans des tubes en téflon.
Deux solutions de spike (solution standard où la concentration en 235U et de 230Th est connue) en 235U (57,41 ppm) et en 230Th (9,35 ppm) sont préalablement diluées dans l’HNO3 5M afin d’obtenir une solution unique de spike possédant une teneur de 0,2032 ppb d’235U et 0,2097 ppb de 230Th. Les cristaux sont dissouts dans 50 µl de la solution acide spikée. La dissolution complète des échantillons nécessite un temps de chauffe pendant deux heures à une température de 90°C. 1 ml d’eau EMQ (eau distillée débarrassée de tout agent organique et particulaire) est rajouté à température ambiante.
A1.3.3.2. Analyse de l’uranium et du thorium
Le contenu de la solution ainsi obtenu est analysé à partir d’un ICP-MS (Induced Coupled Plasma Mass Spectrometer). Les analyses ont été réalisées au LMTG de Toulouse et au LSCE de Gif-sur-Yvette. Le spectromètre de masse associé à un plasma inductif d’argon quadripolaire (X seriesII : Fig. A1-18) est utilisé dans cette étude et va permettre la détection ainsi que le dosage de quantités très faibles d’U et de Th.

Le principe d’analyse est quasi identique par rapport à la spectrométrie à source gazeuse, cependant l’étape d’extraction des éléments à analyser diffère. Le couplage torche à plasma-spectrométrie de masse est une technique permettant de doser des éléments non gazeux à des teneurs très inférieures à 1 µg/l. Cette technique de spectrométrie est basée sur le couplage d'une torche à plasma génératrice d’ions et un spectromètre de masse pour séparer ces ions en masse. Le principe de l’installation est présenté dans la figure suivante (Fig. A1-19).
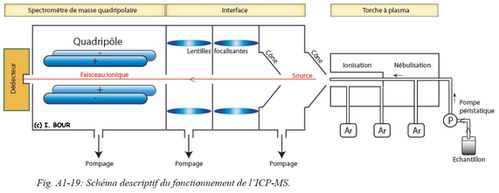
L'échantillon préalablement dissout dans une solution acide est amené jusqu'à la torche à plasma à couplage inductif (ICP) par une pompe péristaltique ou par un effet Venturi. Au contact avec l'argon, l'échantillon est alors nébulisé. L’argon gazeux porté à haute température (plasma de 8000 à 10000 K : Fig. A1-19) est fortement ionisé, puis décompose la matrice, atomise et ionise les espèces introduites dans leur totalité sous forme de cations monovalents.
Une interface composée de deux cônes de nickel (un cône échantillonneur et un cône écrêteur) assure le transfert des ions de l’ICP, partie à pression atmosphérique, vers le quadrupôle, la partie à pression réduite. Un jeu de lentilles électrostatiques, placé après les cônes, permet de refocaliser le faisceau d’ions, qui diverge fortement du fait du gradient de pression, et d’éliminer les espèces neutres ainsi que les photons. Les ions sont focalisés vers le spectromètre de masse (MS) permettant de détecter et quantifier les ions après que ces derniers soient discriminés en fonction de leur rapport masse/charge grâce à l’analyseur. Le faisceau ionique est amené sur un détecteur de type multiplicateur d'électrons. Les isotopes d’238U et de 232Th sont mesurés par comparaison avec le spike introduit.
La réponse des différents éléments chimiques dépend grandement de la température du plasma, de la densité des ions, atomes et électrons dans le plasma, ainsi que l’énergie d’ionisation de ces éléments.
Plusieurs échantillons étalons (protocole selon Evans et al., 2005) sont nécessaires pour calibrer les mesures du détecteur (Tab. A1-1). Un blanc est réalisé au début de chaque série de mesures pour déterminer le bruit de fond. Un blanc spiké est également réalisé pour déterminer les rapports 235U/238U et 230Th/232Th. La mesure d’un standard spiké (mélange d’une solution de spike et d’une solution standard) et d’une solution d’uranium diluée (Analab®) est effectuée tous les 15 échantillons. Le standard spiké permet de vérifier si le rapport 230Th/232Th est reproductible entre les analyses. La solution diluée en uranium renseigne sur le rapport isotopique en uranium (RU) contenu dans un minéral avec RUmin= (235U/238U)min (Evans et al., 2005).

Les différentes parties de la ligne où sont véhiculées les solutions analysées sont rincées par un mélange acide (HCl 3M + HNO3 3M) entre chaque échantillon.
A1.3.4. Reproductibilité des mesures
Pour tester la reproductibilité des mesures effectuées sur l’ensemble des échantillons, il est nécessaire de vérifier l’homogénéité des âges (U-Th)/He obtenus à partir de cristaux d’apatite standards (Fig. A1-20) caractérisées par un âge connu (un âge jeune inférieur à 8 Ma et un âge vieux supérieur à 110 Ma). Ces apatites étalons sont analysées après chaque série de mesures des concentrations He et (U-Th) sur les échantillons. Dans cette étude, les âges (U-Th)/He entre les aliquotes d’échantillons standards sont reproductibles malgré leur FT différent. Cette reproductibilité des mesures atteste que les critères et la qualité de sélection des grains ainsi que le calcul du FT sont corrects. La reproductibilité des âges indique également qu’il n’y a pas de dérive analytique et que la qualité des mesures reste stable au cours du temps.

A1.4. Observation par cathodoluminescence
L'observation en cathodoluminescence est complémentaire de l’observation en microscopie optique standard. C’est une technique d’observation largement utilisée pour les études minéralogiques et pétrographiques (Pagel et al., 2001). En lame mince, la reconnaissance des apatites en lumière naturelle et polarisée n’est pas aisée car elles se confondent facilement avec d’autres minéraux. L’utilisation de la cathodoluminescence présente l’avantage de repérer rapidement la présence de cristaux d’apatite et permet d’estimer leur taille ainsi que leur abondance. Par ce procédé, il est possible de savoir si l’échantillon est exploitable. Une préparation de l’échantillon sous forme de lame mince polie convient pour l’observation en cathodoluminescence. Une épaisseur de 30 µm est adaptée pour l’obtention d’une bonne qualité d’observation ainsi qu’un rendu photographique convenable.
La cathodoluminescence correspond à une radiation lumineuse visible qu'émet un cristal soumis au bombardement d’un faisceau d'électrons allant de quelques keV à une vingtaine de keV. Le bombardement d’électrons a pour effet de générer le passage à un état excité des atomes via l’absorption d’énergie liée à la capture d’électron. L’émission de lumière est la conséquence d’une désexcitation radiative correspondant à un retour à un niveau d’énergie plus faible. Cette émission de lumière est caractérisée par sa couleur et par l'intensité lumineuse des différentes longueurs d'onde qui la composent. Elle dépend notamment des concentrations en éléments traces et de leur environnement électronique dans le cristal (Barbarand et Pagel, 2001).
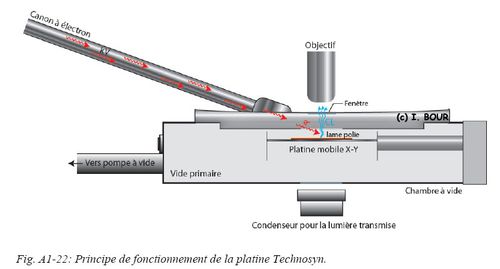
Le système d’appareillage de cathodoluminescence à cathode froide de type Technosyn Mark II est monté sur un microscope optique Nikon (Fig. A1-21). Le montage est équipé d’un générateur de haute tension relié à un canon à électrons oblique où ce dernier est connecté à une chambre à vide dans laquelle est disposé l’échantillon (Fig. A1-22). La chambre à vide est installée au niveau de la platine porte échantillons du microscope. Les réglages des tensions d’accélération des électrons et du vide sont ajustés pour une plage d'utilisation comprise entre 15 et 20 kV avec un vide primaire optimal de 0,8 Torr. En fonction de la stabilité du vide, le courant est ajusté entre 500 et 600 µA. Ces paramètres permettent une bonne stabilité de la luminescence lors de l’observation.

Sous observation par cathodoluminescence, les cristaux d’apatite apparaissent avec une teinte jaune orangée vive (Fig. A1-23.A) qui se distingue nettement des autres teintes plus ternes, notamment dans les textures granitiques. En lumière naturelle, la distinction des cristaux d’apatite n’est pas aisée et rapide (Fig. A1-23.B). La localisation préférentielle des apatites (habitus) au sein de la texture et de la paragenèse de la roche peut ainsi être observées.

A1.5. Analyse à la microsonde électronique
Cette technique est basée sur la spectrométrie des rayons X émis par un échantillon sous l'impact d'un faisceau incident d'électrons. Cette technique a été utilisé sur les échantillons d’Ardenne et de Bohême présentant des valeurs de Dpar extrêmes ainsi que des différences d’âges TF-(U-Th)/He significatives. Ce type d’analyses a pour intérêt d’identifier des caractéristiques chimiques particulières au sein des cristaux d’apatites collectés.
Lorsqu’un faisceau d’électrons bombarde une cible, il se produit dans la matière une diversité d’interactions entre les électrons et les atomes de la cible qui conduisent à l’émission de signaux caractéristiques, électroniques et électromagnétiques exploités en analyse et microanalyse X, en microanalyse Auger, en microscopie électronique à balayage ou en transmission, en spectroscopie X, UV, visible. On admet généralement que le domaine d’interaction du faisceau avec la matière est un volume hémisphérique (Fig. A1-24). Ce volume d’interaction est la source d’émission électronique et électromagnétique : les électrons Auger, les électrons secondaires, les électrons rétrodiffusés, les rayons X, le rayonnement de fluorescence.
La microsonde électronique analyse l’émission X produite par l’interaction entre des électrons incidents et les éléments constituant le matériau à analyser. L’intérêt est d'obtenir la composition chimique du matériau. Cette technique de mesure non destructive permet l'analyse qualitative et quantitative dans un très petit volume de matière, de l'ordre du µm3. La limite de détection est d'environ 10-14 à 10-15 g, ce qui correspond à une teneur de l'ordre de 100 ppm. La microsonde permet l'analyse de tous les éléments de nombre atomique supérieur à 3 (Lithium). L’émission X est appelée émission caractéristique car l’énergie des raies est spécifique à l’élément émettant. L’énergie d’une raie X caractéristique augmente régulièrement avec le numéro atomique de l’atome diffusant.
Le volume analysé est proportionnel à la tension d’accélération, et en première approximation, le volume excité double tous les 5 keV. La sensibilité est affinée en augmentant la tension, mais la résolution spatiale sera réduite. L’intensité du faisceau électronique doit être suffisante pour que le signal X soit statistiquement significatif. L’augmentation de l’intensité électronique permet d’augmenter le diamètre de la sonde mais diminue la résolution.
Pour l’analyse quantitative il est nécessaire d’avoir un échantillon poli optiquement. Une surface rugueuse ou présentant des défauts à l’échelle microscopique (défauts de planéité, cassures, clivage, etch pit, …) entraîne des modifications importantes de l’émission X primaire et donc les concentrations calculées.

Les analyses ont été réalisées au Laboratoire Camparis de l’Université de Paris VI à partir de la microsonde électronique Cameca de type SX100. Le faisceau d’électrons est généré avec une tension de 15 keV et avec une intensité de 10 nA. Le diamètre du faisceau d’électrons incident est de 15 µm. La métallisation des échantillons, nécessaire pour la conduction des électrons, est réalisée en carbone. Les éléments analysés sur les apatites sont le F, Na, Si, P, Cl, Ca, Mn, Fe, Sr, Nd, Sm, et La. Les capteurs de la microsonde sont calibrés à partir de minéraux standards dont les teneurs chimiques sont connues (Tab. A1-2).

Deux points de mesure sont effectués pour chaque grain et 4 à 10 grains par échantillon ont été analysés. Les temps de comptage pour les éléments majeurs (F, Na, Si, P, Cl, Ca, Mn, Fe) sont fixés à 30 secondes et à 10 secondes pour les éléments traces et les éléments de terres rares (Sr, Nd, Sm, La).
A1.6. Observation et analyse au microscopie électronique à balayage
La Microscopie Electronique à Balayage (MEB) est une technique de microscopie électronique, basée sur le principe des interactions électrons-matière, capable de produire des images en haute résolution de la surface d’un échantillon. Cette technique est employée dans ce travail afin d’identifier et de caractériser les inclusions minérales et/ou vitreuses présentent dans la majorité des cristaux d’apatite des cinérites ardennaises et des granitoïdes bohémiens.
Les interactions électrons/matière créent des ionisations au niveau de la surface de l'échantillon. La quantité d'électrons émise est liée à la topographie de la surface de l'échantillon et aussi à sa composition. Le balayage de la surface de l'échantillon par le faisceau d'électrons fourni une image de cette surface qui est reconstituée pixel par pixel dont la valeur des niveaux de gris correspond à l'intensité collectée par le détecteur d'électrons. La surface de l’échantillon est balayée par un faisceau focalisé d’électrons qui va interagir avec la matière (rayonnement primaire). Ces interactions électrons-matière découlent de nombreux rayonnements secondaires (dont l’émission d’électrons secondaires et d’électrons rétrodiffusés) qui sont exploités à l’aide de détecteurs appropriés. Les électrons (secondaires et rétrodiffusés) sont collectés par des détecteurs spécifiques, en synchronisant la détection (intensité) au balayage du faisceau incident. On obtient ainsi une image de la surface dont le contraste est fonction du type d’électrons sélectionnés via le détecteur, de la tension d’accélération choisie, de la nature des atomes présents dans l’échantillon.
Les observations et analyses ont été réalisées au laboratoire IDES (Université Paris XI) à partir du MEB Philips XL serie. La métallisation des échantillons est réalisée en carbone. Les observations MEB dans ce travail sont réalisées par imagerie d’électrons rétrodiffusés avec une tension entre 15 et 30 kV. Ce type d’électron résulte de l’interaction des électrons du faisceau primaire avec des noyaux d’atomes de l’échantillon et qui ont réagi de façon quasi élastique avec les atomes de l’échantillon. La résolution atteinte avec les électrons rétrodiffusés est relativement faible, de l’ordre du micromètre ou du dixième de micromètre mais ces électrons sont sensibles au numéro atomique des atomes constituant l’échantillon. Les atomes les plus lourds (ceux ayant un nombre important de protons) réémettront plus d’électrons que les atomes plus légers. La quantité d’électrons capturés par les atomes rencontrés et donc la quantité d’électrons rétrodiffusés qui ressortent dépend de la nature chimique des couches traversées. Le taux d’émission électronique augmente avec le numéro atomique. Ainsi, il est possible d’obtenir un contraste chimique. Les zones formées d’atomes avec un nombre atomique (Z) élevé apparaîtront plus brillantes que d’autres, c’est le contraste de phase. En revanche, le taux d’émission dépend peu du relief, l’image apparaît donc « plate ». Cette particularité est utilisée pour l’analyse en électrons rétrodiffusés. Cette méthode permet de mesurer l’homogénéité chimique d’un échantillon et permet ainsi une analyse qualitative.
Des analyses complémentaires par rapport à la microsonde ont été effectuées au MEB pour identifier les différentes variétés d’inclusions contenues dans les apatites de cette étude. Suite aux interactions du faisceau d’électrons primaires avec les atomes du matériau analysé, sont créés des rayons X (phénomène de désexcitation). L’énergie de ces rayons X est caractéristique des atomes qui les ont émis, d’où la possibilité de réaliser des analyses élémentaires. La collecte de ces rayons X par un détecteur couplée au MEB a permis de compléter la partie imagerie par des analyses chimiques sur éléments majeurs. La tension du faisceau d’électrons généré pour les analyses est de 15 kV avec un diamètre (spot) de 5,9 µm.



/idata%2F4437986%2FIvan01.jpg)
